发表于 2024-08-26
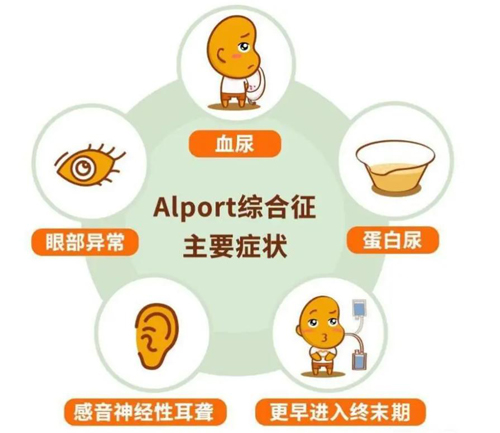
Alport综合征(Alport syndrome,AS)又叫眼-耳-肾综合征,是一种临床表现以血尿、蛋白尿、进行性肾功能减退为特征,部分患者合并感音神经性耳聋、眼部病变等肾外表现的综合征,是导致肾衰竭主要的遗传性肾脏病之一。
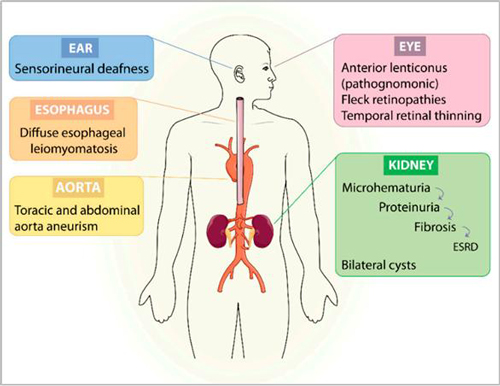
临床表现
1、肾脏表现:
(1)血尿
(2)蛋白尿
(3)终末期肾病
2、听力改变
感音神经性耳聋:初为高频区听力下降,渐及全音域,甚至影响日常的对话交流。
3、眼部病变
(1)前圆锥形晶状体
(2)眼底黄斑周围点状和斑点状视网膜病变
(3)视网膜赤道部视网膜病变
表现为进行性近视,甚至导致前极性白内障或前囊自发穿孔。
4、其他
(1)平滑肌瘤
(2)肌发育不良
(3)甲状腺疾病
(4)AMME综合征(AS伴精神发育迟缓、面中部发育不良及椭圆形红细胞增多症等)。
临床诊断
随着对Alport综合征的认识深入,疾病的诊断标准也经历了几个阶段。自1927年Alport命名后40年,始终以“血尿+耳聋+肾功能衰竭家族史”这一临床综合征标准诊断本病。20世纪70年代电镜技术的应用揭示了本病GBM具有特异性的超微结构改变,在此基础上,Flinter等提出了Alport综合征诊断的4条标准,如果血尿和(或)慢性肾功能衰竭的患者,符合以下4项中的3项便可诊断:
①血尿或慢性肾功能衰竭家族史;
②肾活检电镜检查有典型改变;
③进行性感音神经性耳聋;
④眼部改变;
然而,研究表明,仅45%~55%的Alport综合征患者表现有耳聋,眼部异常的发生率仅为30%~40%,因此上述标准过于严格,会有不少患者漏诊。1996年,Gregory等在综合前人经验的基础上提出诊断Alport综合征的十条标准(见下表)。Alport综合征家系患者诊断:在直系家庭成员中应符合标准中的4条,当然也有例外;但是对于旁系成员及仅表现为不明原因血尿、终末期肾病或听力障碍的个体诊断应十分慎重。判断Alport综合征家族中家族成员是否受累;若该个体符合相应遗传型,再符合标准2-10中一条,可拟诊,符合两条便可确诊。对于无家族史个体的诊断,至少应符合上述指标中4条。
Alport综合征诊断标准:

目前如何治疗Alport综合征?
早期药物对症治疗,延缓ESKD的发生:ACEI/ARB
①血管紧张素转换酶抑制剂(ACEI):雷米普利、赖诺普利等
②血管紧张素Ⅱ受体拮抗剂(ARB):氯沙坦、坎地沙坦等
③其他药物仍处于研究阶段
ESKD:肾脏替代治疗
①血液透析、腹膜透析
②肾移植:成人AS肾移植患者20年存活率为70.2%,移植肾20年存活率为46.8%,均高于其他肾脏病所致的肾移植,且移植肾存活率不受基因变异严重程度影响。
听力障碍:预防为主
①避免暴露于大声喧哗环境
②避免使用耳毒性药物
③出现听力损伤时应用助听器
④助听器效果不佳时可采取人工耳蜗植入术
眼部病变
①护目镜
②前圆锥形晶状体:配镜矫正屈光不正、难以矫正者行晶状体摘除及人工晶体植入术
③角膜糜烂、损伤:局部抗生素、止痛药








